tumor <-readRDS("../test_TransProR/generated_data1/removebatch_SKCM_Skin_TCGA_exp_tumor.rds")normal <-readRDS('../test_TransProR/generated_data1/removebatch_SKCM_Skin_Normal_TCGA_GTEX_count.rds')# Merge the datasets, ensuring both have genes as row namesall_count_exp <-merge(tumor, normal, by ="row.names")all_count_exp <- tibble::column_to_rownames(all_count_exp, var ="Row.names") # Set the row names# Drawing data# all_count_exp <- log_transform(all_count_exp)DEG_deseq2 <-readRDS('../test_TransProR/Select DEGs/DEG_deseq2.Rdata')#head(all_count_exp, 1)head(DEG_deseq2, 5)
# Convert from SYMBOL to ENTREZID for convenient enrichment analysis later. It's crucial to do this now as a direct conversion may result in a reduced set of genes due to non-one-to-one correspondence.# DEG_deseq2# Retrieve gene listgene <-rownames(DEG_deseq2)# Perform conversiongene =bitr(gene, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
'select()' returned 1:many mapping between keys and columns
Warning in bitr(gene, fromType = "SYMBOL", toType = "ENTREZID", OrgDb =
"org.Hs.eg.db"): 43.37% of input gene IDs are fail to map...
# Remove duplicates and mergegene <- dplyr::distinct(gene, SYMBOL, .keep_all=TRUE)# Extract the SYMBOL column as a vector from the first datasetsymbols_vector <- gene$SYMBOL# Use the SYMBOL column to filter corresponding rows from the second dataset by row namesDEG_deseq2 <- DEG_deseq2[rownames(DEG_deseq2) %in% symbols_vector, ]head(DEG_deseq2, 5)
Diff_deseq2 <-filter_diff_genes( DEG_deseq2, p_val_col ="pvalue", log_fc_col ="log2FoldChange",p_val_threshold =0.05, log_fc_threshold =3 )# First, obtain a list of gene names from the row names of the first datasetgene_names <-rownames(Diff_deseq2)# Find the matching rows in the second dataframematched_rows <- all_count_exp[gene_names, ]# Calculate the mean for each rowaverages <-rowMeans(matched_rows, na.rm =TRUE)# Append the averages as a new column to the first dataframeDiff_deseq2$average <- averagesDiff_deseq2$ID <-rownames(Diff_deseq2)Diff_deseq2$changetype <-ifelse(Diff_deseq2$change =='up', 1, -1)# Define a small threshold valuesmall_value <- .Machine$double.xmin# Before calculating -log10, replace zeroes with the small threshold value and assign it to a new columnDiff_deseq2$log_pvalue <-ifelse(Diff_deseq2$pvalue ==0, -log10(small_value), -log10(Diff_deseq2$pvalue))# Extract the expression data corresponding to the differentially expressed genesheatdata_deseq2 <- all_count_exp[rownames(Diff_deseq2), ]#head(heatdata_deseq2, 1)
set.seed(123)# Preparing heatdata for visualizationHeatdataDeseq2 <- TransProR::process_heatdata(heatdata_deseq2, selection =2, custom_names =NULL, num_names_per_group =60, prefix_length =4)HeatdataDeseq2 <-as.matrix(HeatdataDeseq2)library(msigdbr)## Using the msigdbr package to download and prepare for GSVA analysis with KEGG and GO gene sets## KEGGKEGG_df_all <-msigdbr(species ="Homo sapiens", # Homo sapiens or Mus musculuscategory ="C2",subcategory ="CP:KEGG") KEGG_df <- dplyr::select(KEGG_df_all, gs_name, gs_exact_source, gene_symbol)kegg_list <-split(KEGG_df$gene_symbol, KEGG_df$gs_name) # Grouping gene symbols by gs_namehead(kegg_list)
#write.csv(gsva_mat, "gsva_go_matrix.csv")gsva_go <-gsva(expr = HeatdataDeseq2, gset.idx.list = go_list, kcdf ="Poisson", #"Gaussian" for logCPM, logRPKM, logTPM, "Poisson" for countsverbose =TRUE#parallel.sz = parallel::detectCores() # Utilize all available cores )
Warning: Calling gsva(expr=., gset.idx.list=., method=., ...) is deprecated;
use a method-specific parameter object (see '?gsva').
Warning: Some gene sets have size one. Consider setting 'min.sz > 1'.
#write.csv(gsva_mat, "gsva_go_matrix.csv")ssgsea_kegg <-gsva(expr = HeatdataDeseq2, gset.idx.list = kegg_list, kcdf ="Poisson", #"Gaussian" for logCPM, logRPKM, logTPM, "Poisson" for countsmethod ="ssgsea",verbose =TRUE#parallel.sz = parallel::detectCores() # Utilize all available cores)
Warning: Calling gsva(expr=., gset.idx.list=., method=., ...) is deprecated;
use a method-specific parameter object (see '?gsva').
Warning: Some gene sets have size one. Consider setting 'min.sz > 1'.
#write.csv(gsva_mat, "gsva_go_matrix.csv")ssgsea_go <-gsva(expr = HeatdataDeseq2, gset.idx.list = go_list, kcdf ="Poisson", #"Gaussian" for logCPM, logRPKM, logTPM, "Poisson" for countsmethod ="ssgsea",verbose =TRUE#parallel.sz = parallel::detectCores() # Utilize all available cores)
Warning: Calling gsva(expr=., gset.idx.list=., method=., ...) is deprecated;
use a method-specific parameter object (see '?gsva').
Warning: Some gene sets have size one. Consider setting 'min.sz > 1'.
# Data transformation for GSVA of KEGG#### Differential analysis using limma ##### Assigning group labels for tumor and normal samplesgroup <-c(rep('tumor', 60), rep('normal', 60))group <-factor(group, levels =c("normal", "tumor"))# Constructing the design matrixdesign <-model.matrix(~0+factor(group))colnames(design) <-levels(factor(group))rownames(design) <-colnames(gsva_kegg)# Forming the contrast matrix for differential analysiscon <-paste(rev(levels(group)), collapse ="-")cont.matrix <- limma::makeContrasts(contrasts =c(con), levels = design)# Fitting the linear modelfit <- limma::lmFit(gsva_kegg, design)fit2 <- limma::contrasts.fit(fit, cont.matrix)fit2 <- limma::eBayes(fit2)# Retrieving differential expression resultstempOutput <- limma::topTable(fit2, coef = con, n =Inf)gsva_kegg_limma <- stats::na.omit(tempOutput)#### Visualization of differential analysis results from GSVA ###### Setting filtering thresholdspadj_cutoff =0.05log2FC_cutoff =log2(2)# Filtering significant gene setskeep <-rownames(gsva_kegg_limma[gsva_kegg_limma$adj.P.Val < padj_cutoff &abs(gsva_kegg_limma$logFC) > log2FC_cutoff, ])length(keep)
[1] 14
# Extracting the top 10 elements from the limma analysis resultskeep_top10 <- keep[1:10]# Selecting rows in gsva_kegg that match the top 10 genes from keepgsva_kegg_limma_head10 <- gsva_kegg[rownames(gsva_kegg) %in% keep_top10, ]# Checking if row names are set, assuming the first column as row names if not setif (is.null(rownames(gsva_kegg_limma_head10))) { gsva_kegg_limma_head10 <- gsva_kegg_limma_head10 %>%mutate(RowID =row_number()) %>%# Adding a row number for each rowcolumn_to_rownames(var="RowID") # Setting this row number as row names}gsva_kegg_limma_head10 <-as.data.frame(gsva_kegg_limma_head10)# To preserve row names in the long format data, first convert row names into a column of the data framegsva_kegg_limma_head10 <- gsva_kegg_limma_head10 %>% tibble::rownames_to_column(var ="Gene") # Convert row names to a column named Gene# Transforming the dataframe from wide to long format, including the newly added row names columngsva_kegg_HeatdataDeseq <- gsva_kegg_limma_head10 %>%pivot_longer(cols =-Gene, # Transforming all columns except the Gene columnnames_to ="Sample", # Creating a new column to store the original column namesvalues_to ="value"# Creating a new column to store the values )# Displaying the transformed dataprint(gsva_kegg_HeatdataDeseq)
# Subsetting the first 10 rows from HeatdataDeseq2HeatdataDeseq3 <- HeatdataDeseq2[1:10,]# Applying logarithmic transformation and converting to a data frameHeatdataDeseq3 <-as.data.frame(log_transform(HeatdataDeseq3))
[1] "log2 transform finished"
# Check if row names are set, if not, assume the first column as row namesif (is.null(rownames(HeatdataDeseq3))) { HeatdataDeseq3 <- HeatdataDeseq3 %>%mutate(RowID =row_number()) %>%# Add a row number to each rowcolumn_to_rownames(var="RowID") # Set this row number as row names}# To retain row names in a long format, first convert row names to a column in the data frameHeatdataDeseq3 <- HeatdataDeseq3 %>% tibble::rownames_to_column(var ="Gene") # Convert row names to a column named 'Gene'# Transforming the data frame from wide to long format, including the newly added row name columnlong_format_HeatdataDeseq <- HeatdataDeseq3 %>%pivot_longer(cols =-Gene, # Transform all columns except for 'Gene'names_to ="Sample", # Create a new column to store original column namesvalues_to ="value"# Create a new column to store values )# Display the transformed dataprint(long_format_HeatdataDeseq)
# Selecting the top 10 rows from ssgsea_kegg for displayssgsea_kegg_head10 <-as.data.frame(ssgsea_kegg[1:10,])# Check if row names are set, if not, assume the first column as row namesif (is.null(rownames(ssgsea_kegg_head10))) { ssgsea_kegg_head10 <- ssgsea_kegg_head10 %>%mutate(RowID =row_number()) %>%# Add a row number to each rowcolumn_to_rownames(var="RowID") # Set this row number as row names}ssgsea_kegg_head10 <-as.data.frame(ssgsea_kegg_head10)# To preserve row names in the long format data, first convert row names into a column of the data framessgsea_kegg_head10 <- ssgsea_kegg_head10 %>% tibble::rownames_to_column(var ="Gene") # Convert row names to a column named Gene# Transforming the data frame from wide to long format, including the newly added row name columnssgsea_kegg_HeatdataDeseq <- ssgsea_kegg_head10 %>%pivot_longer(cols =-Gene, # Transform all columns except for 'Gene'names_to ="Sample", # Create a new column to store original column namesvalues_to ="value"# Create a new column to store values )# Display the transformed dataprint(ssgsea_kegg_HeatdataDeseq)
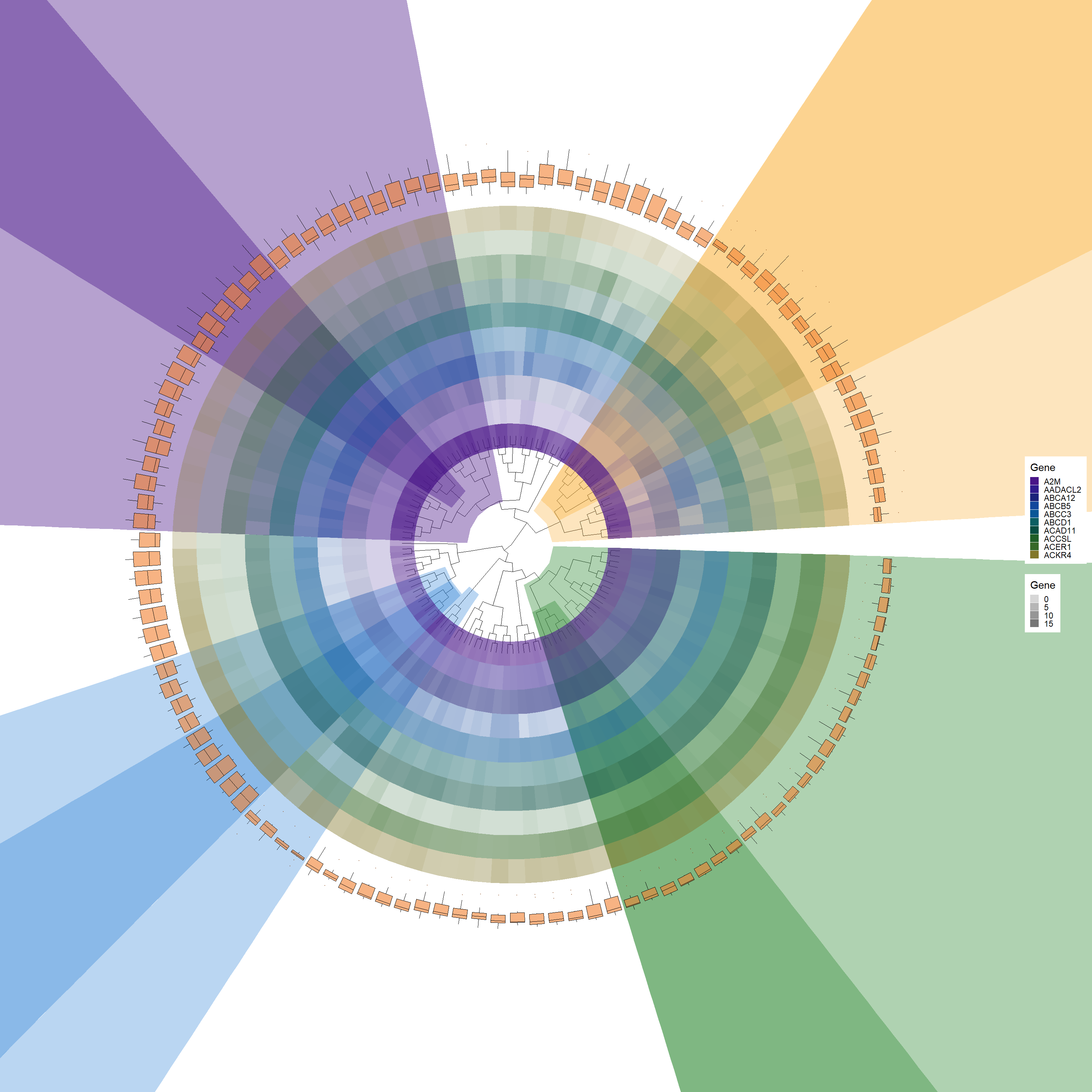
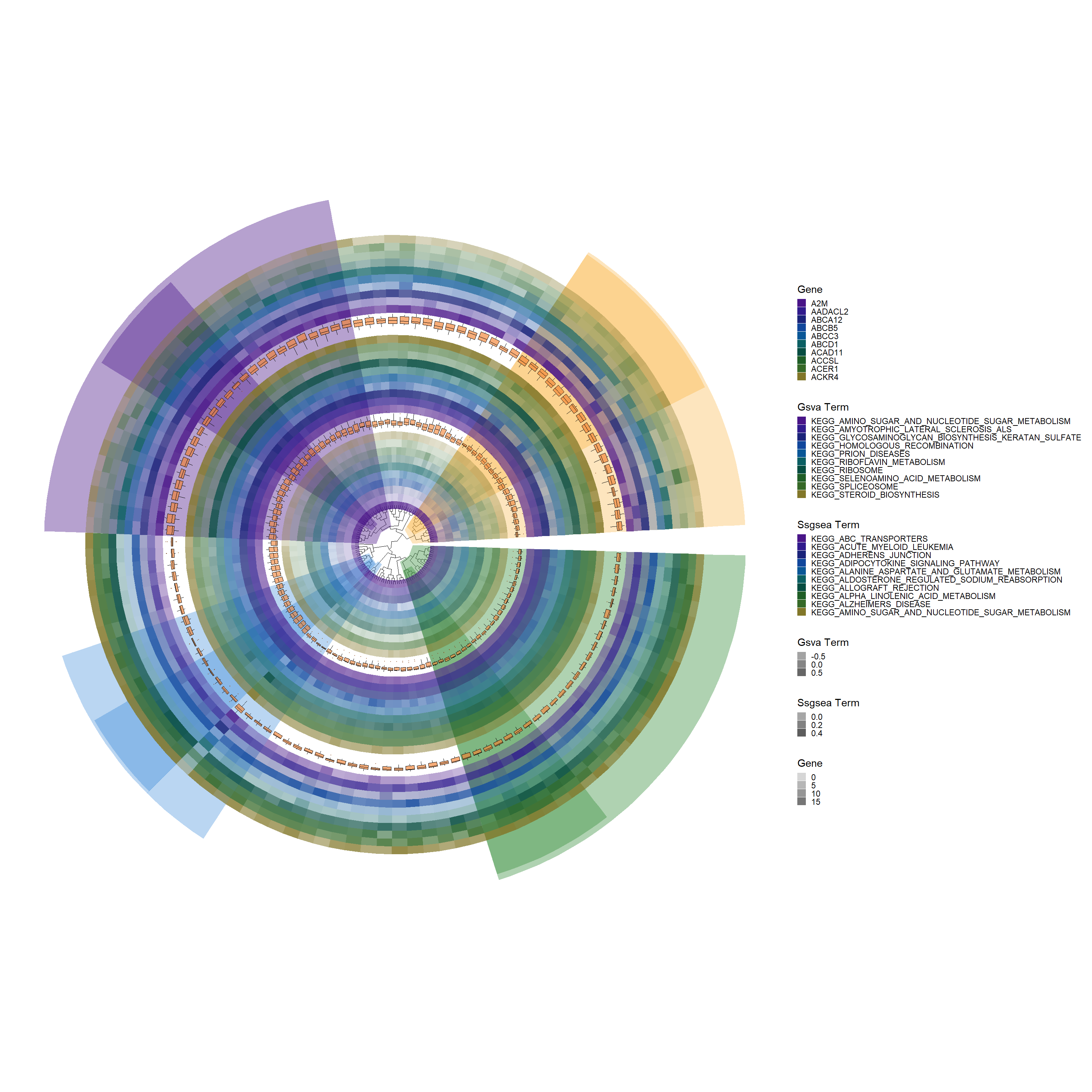
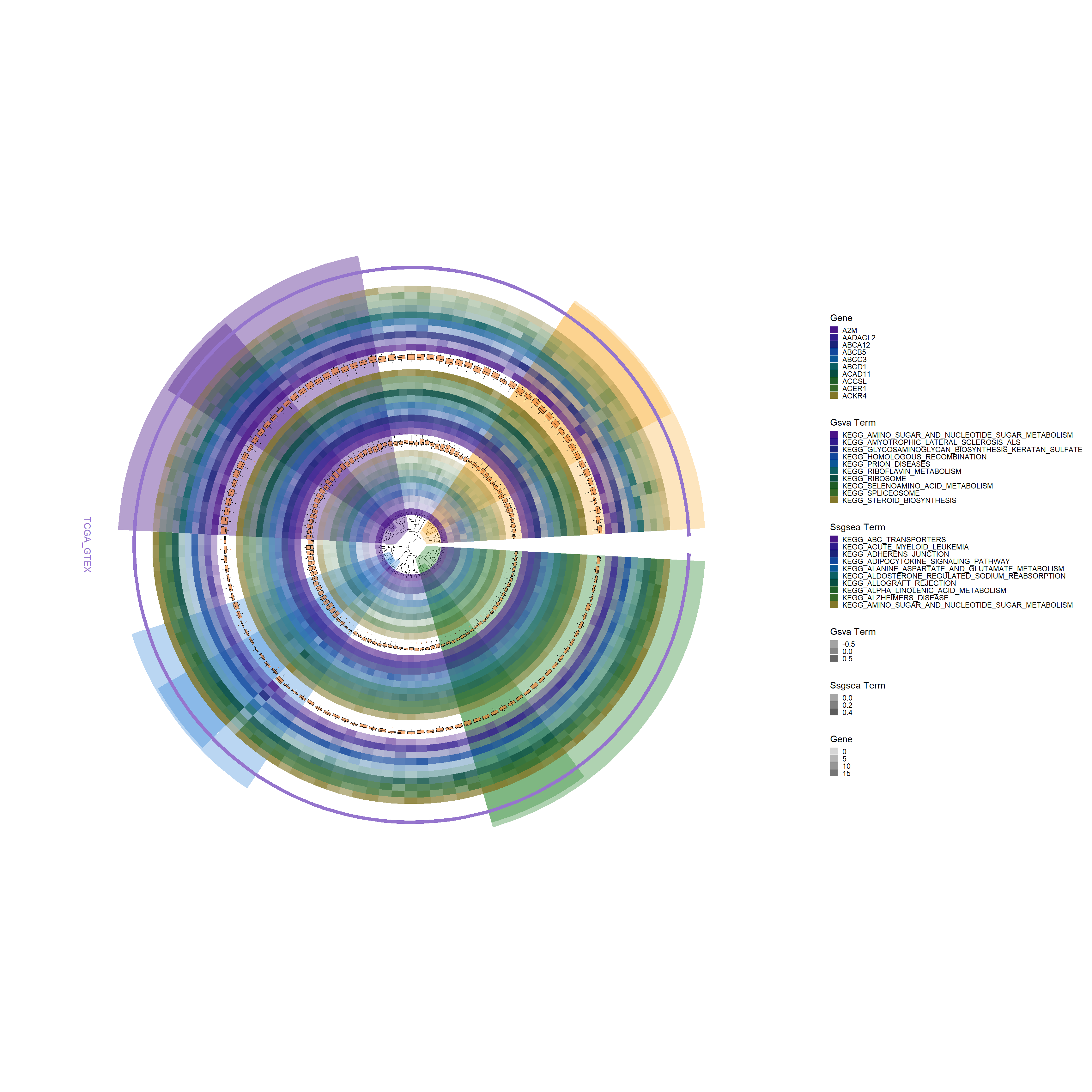
# Add a boxplot layer to the base plotp3 <-add_boxplot(p2, long_format_HeatdataDeseq)p3
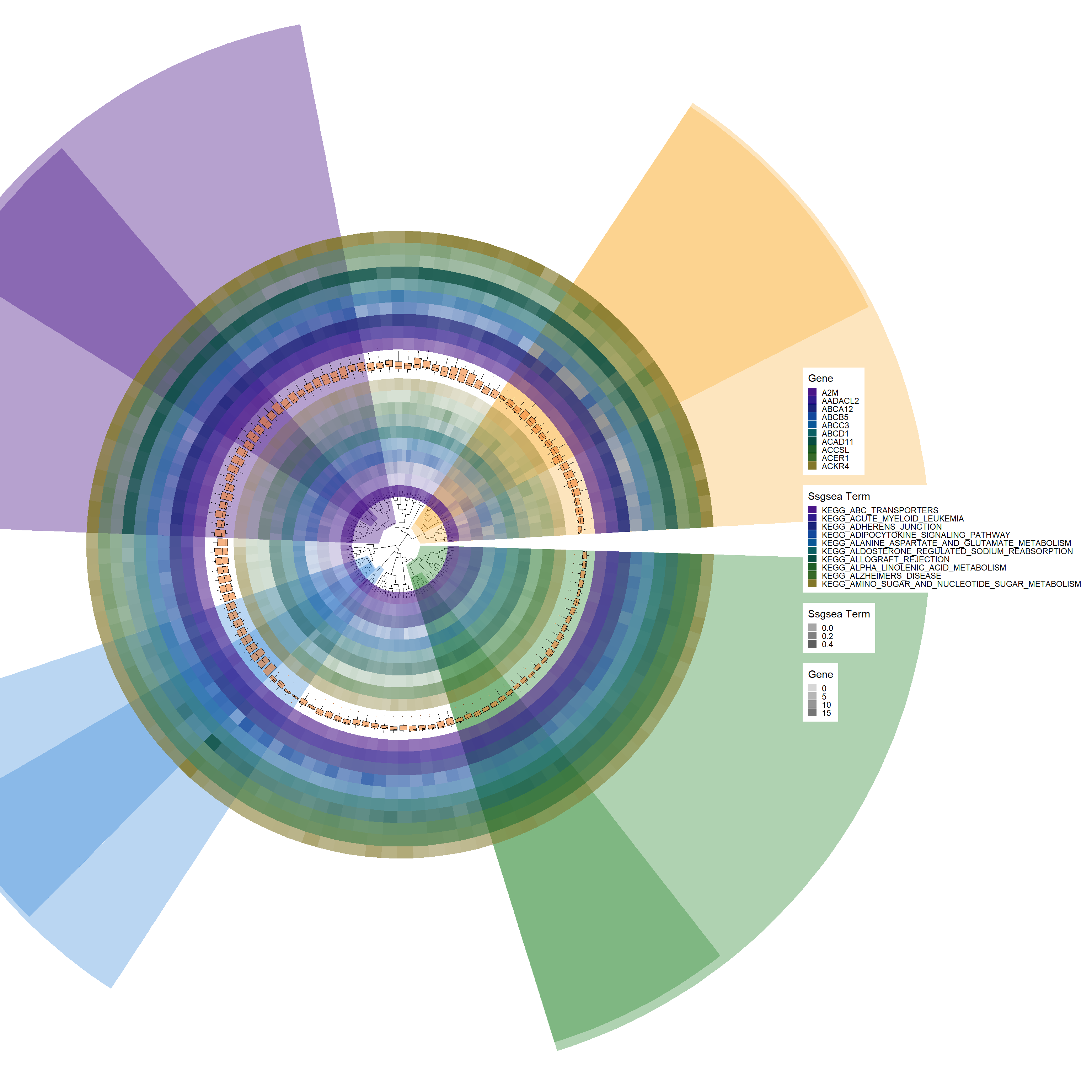
gene_colors <-c("#491588", "#301b8d", "#1a237a", "#11479c", "#0a5797","#0b5f63","#074d41","#1f5e27","#366928","#827729")# Add a new tile layer for SSGSEA datap4 <-add_new_tile_layer(p3, ssgsea_kegg_HeatdataDeseq, gene_colors, "Ssgsea Term")p4
# Add another boxplot layer with specific aesthetic adjustmentsp5 <-add_boxplot(p4, ssgsea_kegg_HeatdataDeseq, fill_color="#f28131", alpha=0.65, offset=0.32)p5
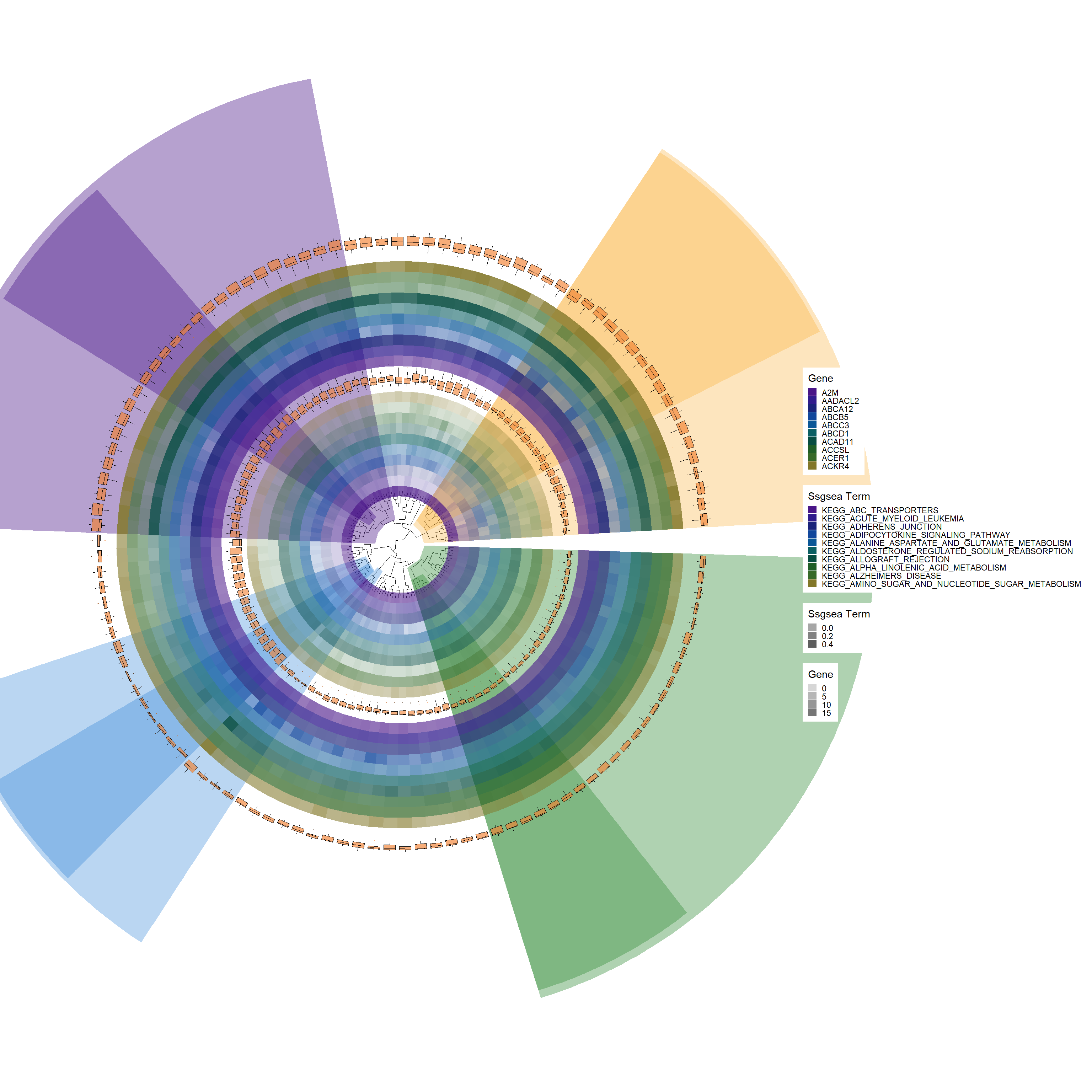
gene_colors <-c("#491588", "#301b8d", "#1a237a", "#11479c", "#0a5797","#0b5f63","#074d41","#1f5e27","#366928","#827729")# Add a new tile layer for GSVA data with specific alpha and offset adjustmentsp6 <-add_new_tile_layer(p5, gsva_kegg_HeatdataDeseq, gene_colors, "Gsva Term", alpha_value=c(0.3, 0.9), offset=0.02)p6